
CRISPR/Casによるノックイン Q&A
CRISPR/Casによるゲノム編集の優れたアプリケーションのひとつに、相同組換修復(HDR)経路を介して特定のゲノム遺伝子座にDNA配列を正確に挿入、あるいは置換するノックイン法がある。部位特異的なノックインを行うこの手法はますます注目が集まっているが、その方法は多岐にわたり、対象となる生物や細胞の種類、ゲノム上での遺伝子座、ドナーDNAなどの要因により、その効率は大きく異なる。
相同組換修復によるゲノム編集ノックインの成功のため、よくある質問に対する回答を以下にまとめた。
相同組換修復によるゲノム編集ノックインの成功のため、よくある質問に対する回答を以下にまとめた。
全表示/全非表示
Q1 どのようにしてノックイン用ドナーDNAを設計すれば良いか?
A1 ドナーDNAの配列は、ノックインしたい目的配列の5’末端および3’末端に、野生型ゲノム遺伝子座と相同な配列を持たせる必要があります。これらの5’および3’配列は、「ホモロジーアーム」と呼ばれます。
ノックインしたい目的配列は、ドナーDNAの中央部に配置する必要があります。ドナーDNAの配列は、sgRNAとCas9ヌクレアーゼによる切断箇所とは無関係ですが、効率的なノックインを行うためには、挿入位置がCas9-sgRNA複合体(RNP)による切断箇所にできるだけ近い位置にあることが望ましいです(詳細は以下のFAQをご参照ください)。
下図は、各ゲノム遺伝子座において、一塩基置換もしくは長鎖インサートの挿入をする際のドナーDNA(HDR template)をそれぞれ示しています。

図1. 配列の1塩基置換または挿入を行うためのノックイン用ドナーDNA構造例
いずれの場合も、ドナーDNAは、目的配列が野生型の標的部位と相同的な5’および3’配列に挟まれている必要がある。
パネルA:RECQL4遺伝子の1,488番目のCをTに置換するために設計したドナーDNA。置換した塩基は赤で示されている。
パネルB:UGT1A9遺伝子のC末端にmycタグを挿入するために設計したドナーDNA。今回は標的配列の中で、挿入を行いたい位置に比較的近い配列に対して異なる3つのsgRNAを設計した。
ノックインしたい目的配列は、ドナーDNAの中央部に配置する必要があります。ドナーDNAの配列は、sgRNAとCas9ヌクレアーゼによる切断箇所とは無関係ですが、効率的なノックインを行うためには、挿入位置がCas9-sgRNA複合体(RNP)による切断箇所にできるだけ近い位置にあることが望ましいです(詳細は以下のFAQをご参照ください)。
下図は、各ゲノム遺伝子座において、一塩基置換もしくは長鎖インサートの挿入をする際のドナーDNA(HDR template)をそれぞれ示しています。
図1. 配列の1塩基置換または挿入を行うためのノックイン用ドナーDNA構造例
いずれの場合も、ドナーDNAは、目的配列が野生型の標的部位と相同的な5’および3’配列に挟まれている必要がある。
パネルA:RECQL4遺伝子の1,488番目のCをTに置換するために設計したドナーDNA。置換した塩基は赤で示されている。
パネルB:UGT1A9遺伝子のC末端にmycタグを挿入するために設計したドナーDNA。今回は標的配列の中で、挿入を行いたい位置に比較的近い配列に対して異なる3つのsgRNAを設計した。
Q2 ノックインを行う際、二本鎖ではなく一本鎖のドナーDNAを使用するメリットはあるか?
A2 一本鎖DNA(ssDNA)を使用すると、二本鎖DNA(dsDNA)に比べて細胞毒性が低く、ランダムインテグレーションの頻度が減ることが実証されており(Roth et al. 2018; Li et al. 2017)、この利点は、遺伝子導入が難しい細胞株を扱う場合において、特に重要となります。例えば、標的細胞で発現する内在性遺伝子に蛍光タンパク質を融合させる場合、ゲノム編集を行った細胞集団内において蛍光を示す細胞の割合は、目的ノックインの成功率と言い換えることができます。これらの蛍光細胞の一部は標的外の遺伝子にドナーDNAがランダムインテグレーションによる相同組換が起きたことが原因で蛍光を示している可能性がありますが、ssDNAをドナーDNAとして用いた場合には発生しにくくなります。
参考文献
Design and specificity of long ssDNA donors for CRISPR-based knock-in. Li, H et al., bioRxiv (2017) doi: https://doi.org/10.1101/178905.
Reprogramming human T cell function and specificity with non-viral genome targeting. Roth, T.L et al., Nat. Lett. (2018) 559, 405–409.
参考文献
Design and specificity of long ssDNA donors for CRISPR-based knock-in. Li, H et al., bioRxiv (2017) doi: https://doi.org/10.1101/178905.
Reprogramming human T cell function and specificity with non-viral genome targeting. Roth, T.L et al., Nat. Lett. (2018) 559, 405–409.
Q3 一本鎖オリゴデオキシヌクレオチド(ssODN)は一本鎖DNA(ssDNA)とどう違うのか?また、ドナーDNAとして、どのような場合にどちらを使うのがよいか?
A3 ssODNは、通常、化学合成によって作製され、最大で約200 ntのドナーDNAとして、小規模なゲノム編集(例: 一塩基置換や50 nt以下の短い挿入)を行う際によく使用されます。500 nt以上のドナーDNAを必要とするような長い挿入を行う場合には、Guide-it Long ssDNA Production System v2(製品コード 632666)を使用して調製したssDNAの使用をお勧めします。
Q4 ドナーDNAとしてssODNやssDNAを使用する場合、ゲノム編集の標的領域に対してセンス鎖、アンチセンス鎖のどちらの使用が適しているか?
A4 200 nt以上のドナーDNAでは現在、センス鎖、アンチセンス鎖のどちらかを優先的に使用するかについて明確な知見はありません。しかし、より短鎖のssODNの場合は、その方向性(センス鎖を使うのか、アンチセンス鎖を使うのか)が、効率に影響することが分かっています(Bollen et al.2018)。
参考文献
How to create state-of-the-art genetic model systems: strategies for optimal CRISPR-mediated genome editing. Bollen, Y., Post, J., Koo, B. & Snippert, H.J.G Nuc. Acid. Res. (2018) 46, 6435–6454.
参考文献
How to create state-of-the-art genetic model systems: strategies for optimal CRISPR-mediated genome editing. Bollen, Y., Post, J., Koo, B. & Snippert, H.J.G Nuc. Acid. Res. (2018) 46, 6435–6454.
Q5 ドナーDNAのホモロジーアームの最適な長さはどれくらいか?
A5 ホモロジーアームの長さとノックイン効率の間には指数関数的な関係があり、350~700 ntの長さのホモロジーアームを含むssDNAテンプレートが最適なパフォーマンスを提供することが実証されています(Li et al. 2017)。ヒトiPS細胞を用いた我々の研究では、350 ntのホモロジーアームで最適なパフォーマンスが観察され、より長いアームを使用してもノックイン効率は大幅に増加することはありませんでした。
長いホモロジーアームを使用すると、ドナーDNAの分子量が増加します。エレクトロポレーションに使用するドナーDNAの量は通常μg単位で測定されるため、ホモロジーアームが長いと、標的細胞に導入されるドナーDNAのモル(コピー)数が少なくなり、ノックイン効率に影響を与える可能性があることを留意しなければなりません。
参考文献
Design and specificity of long ssDNA donors for CRISPR-based knock-in. Li, H et al., bioRxiv (2017) doi: https://doi.org/10.1101/178905.
長いホモロジーアームを使用すると、ドナーDNAの分子量が増加します。エレクトロポレーションに使用するドナーDNAの量は通常μg単位で測定されるため、ホモロジーアームが長いと、標的細胞に導入されるドナーDNAのモル(コピー)数が少なくなり、ノックイン効率に影響を与える可能性があることを留意しなければなりません。
参考文献
Design and specificity of long ssDNA donors for CRISPR-based knock-in. Li, H et al., bioRxiv (2017) doi: https://doi.org/10.1101/178905.
Q6 PAM配列やsgRNAの認識領域にサイレント変異を導入したドナーDNAを使用することは有効か?
A6 Cas9-sgRNA RNPは、プロトスペーサー配列中のPAM配列、もしくはsgRNA認識領域の変更を伴わないノックインの場合、ゲノム編集後もターゲット領域が再切断されて、せっかく目的のノックインが起こっていても再度そこがゲノム編集されてしまう可能性があります。そこで、ノックインの成功率を高めるための一般的な対策として、ドナーDNAにサイレント変異を導入し、対応するアミノ酸配列を変えることなくCas9-sgRNA RNPの再結合・再切断を阻害する方法があります。
PAM配列がコード領域にあり、サイレント変異が可能な場合は、ゲノム編集の効率を最大限に高め、ノックイン後のCas9による再切断やインデルの発生を防ぐために、サイレント変異を入れることが望ましいです。(Paquet et al. 2016)
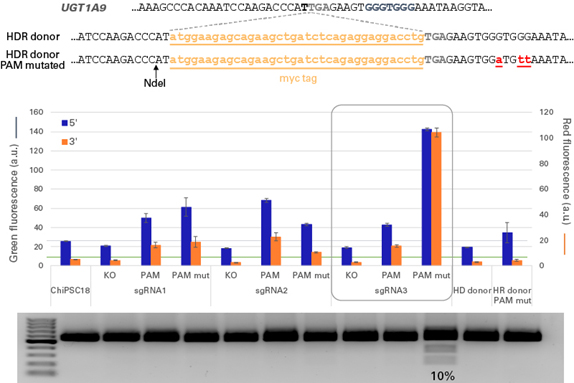
図2. PAM配列の変異によるゲノム編集効率の向上
CRISPR/Casによって、UGT1A9遺伝子のC末端の終始コドン(TGA、グレー表記)の直前にmycタグ(黄色表記、小文字の塩基配列)を挿入するノックイン実験を行った。挿入部近傍の配列に対して3種類のsgRNA(sgRNA1、sgRNA2、sgRNA3)を設計し、それぞれテストに用いた。また、ドナーDNAとして、異なる2つの配列(1つは一切変異がないもの;HRD donor、もう1つは3種類のsgRNAに対応するPAMサイトに変異を持つもの;HRD donor PAM mutated)を用いた。これらを組み合わせてCas9-SgRNA複合体(RNP)を形成させ、エレクトロポレーションによってChiPSC18細胞に導入した。エレクトロポレーション後は、Guide-it SNP Screening Kitを用いて挿入配列の5’末端と3’末端をそれぞれ緑色と赤色の蛍光シグナルで検出し、ノックイン効率を解析した。その結果、sgRNA3とPAMサイトに変異を持つドナーDNAを組み合わせた場合に、両方の波長で最も強いシグナルが検出された。さらに、2つのドナーDNAはゲノムに挿入されると新たにNde Iサイトを生成する様に設計されている。これを利用してRFLPベースの方法でもノックイン効率を調べた。その結果、sgRNA3とPAMサイトに変異を持つドナーDNAとの組み合わせでのみシグナルが検出され、Guide-it SNP Screening Kitの結果と一致した。
参考文献
DNA targeting specificity of RNA-guided Cas9 nucleases. Hsu, P. D. et al. Nat. Biotechnol. (2013) 31, 827–32.
Precise and efficient scarless genome editing in stem cells using CORRECT. Kwart, D., Paquet, D., Teo, S. & Tessier-Lavigne, M. Nat. Protoc. (2017) 12, 329–354.
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.
Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Yamano, T. et al. Mol. Cell (2017) 67, 633–645.e3.
Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Zhang, Y. et al. Sci. Rep. (2014) 4, doi: 10.1038/srep05405.
PAM配列がコード領域にあり、サイレント変異が可能な場合は、ゲノム編集の効率を最大限に高め、ノックイン後のCas9による再切断やインデルの発生を防ぐために、サイレント変異を入れることが望ましいです。(Paquet et al. 2016)
図2. PAM配列の変異によるゲノム編集効率の向上
CRISPR/Casによって、UGT1A9遺伝子のC末端の終始コドン(TGA、グレー表記)の直前にmycタグ(黄色表記、小文字の塩基配列)を挿入するノックイン実験を行った。挿入部近傍の配列に対して3種類のsgRNA(sgRNA1、sgRNA2、sgRNA3)を設計し、それぞれテストに用いた。また、ドナーDNAとして、異なる2つの配列(1つは一切変異がないもの;HRD donor、もう1つは3種類のsgRNAに対応するPAMサイトに変異を持つもの;HRD donor PAM mutated)を用いた。これらを組み合わせてCas9-SgRNA複合体(RNP)を形成させ、エレクトロポレーションによってChiPSC18細胞に導入した。エレクトロポレーション後は、Guide-it SNP Screening Kitを用いて挿入配列の5’末端と3’末端をそれぞれ緑色と赤色の蛍光シグナルで検出し、ノックイン効率を解析した。その結果、sgRNA3とPAMサイトに変異を持つドナーDNAを組み合わせた場合に、両方の波長で最も強いシグナルが検出された。さらに、2つのドナーDNAはゲノムに挿入されると新たにNde Iサイトを生成する様に設計されている。これを利用してRFLPベースの方法でもノックイン効率を調べた。その結果、sgRNA3とPAMサイトに変異を持つドナーDNAとの組み合わせでのみシグナルが検出され、Guide-it SNP Screening Kitの結果と一致した。
参考文献
DNA targeting specificity of RNA-guided Cas9 nucleases. Hsu, P. D. et al. Nat. Biotechnol. (2013) 31, 827–32.
Precise and efficient scarless genome editing in stem cells using CORRECT. Kwart, D., Paquet, D., Teo, S. & Tessier-Lavigne, M. Nat. Protoc. (2017) 12, 329–354.
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.
Structural Basis for the Canonical and Non-canonical PAM Recognition by CRISPR-Cpf1. Yamano, T. et al. Mol. Cell (2017) 67, 633–645.e3.
Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Zhang, Y. et al. Sci. Rep. (2014) 4, doi: 10.1038/srep05405.
Q7 ノックインにおける挿入部位は、PAM配列と隣接している必要があるか?
A7 ノックインにおける挿入部位は、Cas9-sgRNAのRNP切断部位の上流または下流の10 nt以内に配置することが強く推奨されています。挿入部位からCas9-sgRNA RNP切断部位までの距離と、ノックイン効率との間には逆相関関係(挿入部位と切断部位の距離が長くなればなるほど、ノックイン効率が低下する)があることが実証されています(Paquet et al.)。
参考文献
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.
参考文献
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.
Q8 クロマチン構造がノックインの効率に影響を与えるか?
A8 in vivoにおいて、ヌクレオソームがSpCas9による標的部位の切断を阻害することが示されていることから、sgRNA自身の切断活性に加えて、Cas9の活性がクロマチンの構造によっても影響を受けることが示唆されています(Yarrington et al.2018)。DNA配列は、部位特異的なインデルに対する重要な要因ではありますが、クロマチンへのDNAの折りたたみが、各遺伝子座におけるゲノム編集効率やインデルの効率に影響を与えうることが示唆されています。(Chakrabarti et al.2018)。
参考文献
Target-Specific Precision of CRISPR-Mediated Genome Editing. Chakrabarti, A. M. et al. Mol. Cell (2018) 73, 1–15.
Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Yarrington, R. M., Verma, S., Schwartz, S., Trautman, J. K. & Carroll, D. Proc. Natl. Acad. Sci. (2018) 115, 9351–9358.
参考文献
Target-Specific Precision of CRISPR-Mediated Genome Editing. Chakrabarti, A. M. et al. Mol. Cell (2018) 73, 1–15.
Nucleosomes inhibit target cleavage by CRISPR-Cas9 in vivo. Yarrington, R. M., Verma, S., Schwartz, S., Trautman, J. K. & Carroll, D. Proc. Natl. Acad. Sci. (2018) 115, 9351–9358.
Q9 ノックイン効率を上げるための、他の方法はあるか?
A9 現在、ゲノム編集分野では、ノックイン効率を向上させるための新たな手法の開発が重要な課題となっています。これらの課題のために、以下のようなアプローチが期待されています。
参考文献
Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Aird, E. J., Lovendahl, K. N., St. Martin, A., Harris, R. S. & Gordon, W. R. Commun. Biol. (2018) 1, 54.
Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Carlson-Stevermer, J. et al. Nat. Commun. (2017) 8, 1711.
Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Gutschner, T., Haemmerle, M., Genovese, G., Draetta, G. F. & Chin, L. Cell Rep. (2016) 14, 1555–1566.
Base editing: precision chemistry on the genome and transcriptome of living cells. Rees, H. A. & Liu, D. R. Nat. Rev. Genet. (2018) 19, 770–788.
Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Riesenberg, S. & Maricic, T. Nat. Commun. (2018) 9, 2164.
Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Savic, N. et al. Elife (2018) 7,.
Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Wienert, B. et al. bioRxiv (2018) 500462. doi:10.1101/500462
- Cas9-sgRNA RNPとテンプレートの融合または接合(Aird et al. 2018; Savic et al. 2018; Carlson-Stevermer et al. 2017)
- ノックインが最も起きやすいとされているS期およびG2期におけるCas9の発現を制御(Gutschner et al. 2016)
- 一塩基置換のための塩基編集技術の開発(Rees and Liu 2018)
- 相同組換修復の効率を高める様々な化合物の適用(Wienert et al. 2018; Riesenberg and Maricic 2018)
参考文献
Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Aird, E. J., Lovendahl, K. N., St. Martin, A., Harris, R. S. & Gordon, W. R. Commun. Biol. (2018) 1, 54.
Assembly of CRISPR ribonucleoproteins with biotinylated oligonucleotides via an RNA aptamer for precise gene editing. Carlson-Stevermer, J. et al. Nat. Commun. (2017) 8, 1711.
Post-translational Regulation of Cas9 during G1 Enhances Homology-Directed Repair. Gutschner, T., Haemmerle, M., Genovese, G., Draetta, G. F. & Chin, L. Cell Rep. (2016) 14, 1555–1566.
Base editing: precision chemistry on the genome and transcriptome of living cells. Rees, H. A. & Liu, D. R. Nat. Rev. Genet. (2018) 19, 770–788.
Targeting repair pathways with small molecules increases precise genome editing in pluripotent stem cells. Riesenberg, S. & Maricic, T. Nat. Commun. (2018) 9, 2164.
Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Savic, N. et al. Elife (2018) 7,.
Timed inhibition of CDC7 increases CRISPR-Cas9 mediated templated repair. Wienert, B. et al. bioRxiv (2018) 500462. doi:10.1101/500462
Q10 対立アレルに関して、片方に変異が入り、もう片方は野生型のままのクローンの取得効率を上げるにはどうしたらよいか?
A10 一方のアレルのみが改変され、他方のアレルが編集されていないクローンを得ることは特に困難であると言われています。そのような中、SNP/野生型のヘテロアレルを持つクローンをより効率的に得るために、研究者たちは2つの方法を開発しました(Paquet et al.2016)。
参考文献
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.
- 切断部位が挿入部位から10 nt以上離れたsgRNAを使用する
- 変異型と野生型のノックインドナーDNAを等量混合して使用する(1つはSNPをコードし、もう1つは野生型アレルをコードしたもの。いずれもPAM配列に変異が入るように設計するQ6参照)
参考文献
Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Paquet, D. et al. Nature (2016) 533, 125–129.