
修飾酵素の使用に際して
全表示/全非表示
T4 DNA Ligase
Q1 平滑末端と突出末端におけるライゲーション効率の差は?
A1 末端塩基配列にもよります(下記参照)が、突出末端の方が100倍以上ライゲーションしやすくなります。
【参考】一般的に次のような傾向が見られます。
【参考】一般的に次のような傾向が見られます。
| 突出末端 | : | Hind III>Pst I>EcoR I>BamH I>Sal I (Hind III siteはSal I siteより10~40倍速くライゲーションされる) |
| 平滑末端 | : | Hae III>Alu I>Hinc II>Sma I (Hae III siteはSma I siteより5~10倍速くライゲーションされる) EcoR V>Sca I>Pvu II>Nru I |
Q2 分子内ライゲーションより分子間ライゲーションが起こりやすくなるDNA濃度は?
A2 DNAの大きさにもよりますが、DNA濃度は薄ければ薄いほど分子内ライゲーションが起こりやすくなります。
理論上、計算式:51.1/(DNA濃度:g/l)÷(分子量:dalton)0.5の値が1より小さいと分子間ライゲーションの方が、2より大きいと分子内ライゲーションの方が起こりやすくなります1)。
例)
1) Dugaiczyk, A., Boyer, H. W. and Goodman, H. M.(1975)J. Mol. Biol. 96, 171-184.
理論上、計算式:51.1/(DNA濃度:g/l)÷(分子量:dalton)0.5の値が1より小さいと分子間ライゲーションの方が、2より大きいと分子内ライゲーションの方が起こりやすくなります1)。
例)
| λDNA (48,502 bp) |
→ | 1 μg/100 μlより濃いと分子間ライゲーションの方が起こりやすい。 1 μg/200 μlより薄いと分子内ライゲーションの方が起こりやすい。 |
| プラスミドベクター (約3,000 bp) |
→ | 1 μg/25 μlより濃いと分子間ライゲーションの方が起こりやすい。 1 μg/50 μlより薄いと分子内ライゲーションの方が起こりやすい。 |
1) Dugaiczyk, A., Boyer, H. W. and Goodman, H. M.(1975)J. Mol. Biol. 96, 171-184.
Q3 10%ポリエチレングリコール存在下、トランスフォーメーションをしてもよいのか?
A3 コンピテントセル100 μlあたりライゲーション溶液(10%PEGを含む)は10 μlまでなら大丈夫ですが、できればエタノール沈殿を行いバッファー交換する方が望ましいでしょう。
Q4 DNAを環状にしたいが塩濃度(NaCl、KCl等)はどのくらいがよいか?
A4 環状にしたい(セルフライゲーション)場合は一価の塩を含まないバッファーで行ってください。PEGと高塩濃度で促進されるのは分子間ライゲーションであり、分子内ライゲーションは塩濃度を含まないバッファーで行う方がよい結果が得られます。
Q5 力価表示が、文献値や他社の表示と異なっているが?
A5 タカラバイオのT4 DNA Ligaseは、λ-DNA Hind III分解物のライゲーション効率により力価を定義しているため、ATP-PPi交換反応で測定した力価表示(Weiss Unit)とは異なっています。1 Weiss Unitは、タカラバイオのT4 DNA Ligaseの約125 Uに相当します。
T4 RNA Ligase
Q1 タカラバイオの活性測定法は他社と異なっているが、換算するとどうなるか?
A1 タカラバイオ(3'標識活性)の3 Uは他社(self-circularization活性)の1 Uに相当します。
T4 Polynucleotide Kinase
Q1 DNAをラベルする際、〔γ-32P〕ATPの代わりに〔γ-35S〕ATPは使えるか?
A1 使えます。ただし〔γ-32P〕ATPのときより2~3倍高い酵素濃度が必要です。
Q2 T4 Polynucleotide Kinaseの失活方法は?
A2 70℃、5分の加熱処理で失活します。
Alkaline Phosphatase(BAP、CIAP共通)
Q1 Alkaline Phosphataseによる脱リン酸化処理の効率を高める方法は?
A1 以下のどちらか、あるいは両方の操作で効率を上げることができます。
- 反応温度を上げて反応する。(BAPの場合は約65℃、CIAPの場合は約50℃)
- 反応液中のTris濃度を1 Mまで上げる。
Q2 BAPとCIAPの使い分けは?
A2 BAP、CIAPは熱安定性や失活条件が異なります。BAPは非常に安定性の高い酵素なので、反応終了後少なくとも2回フェノール処理を行なってください。CIAPは、キレート剤存在下65℃、30分の熱処理で99%以上の活性が不可逆的に失活しますが、使用条件によっては不充分な場合もあるので、完全に失活させるためにはフェノール処理を行なってください。
一般に平滑末端や5'陥没末端は脱リン酸化の効率が悪くなります。その場合は、BAPを用いた高温(55~60℃)での反応をおすすめします。
一般に平滑末端や5'陥没末端は脱リン酸化の効率が悪くなります。その場合は、BAPを用いた高温(55~60℃)での反応をおすすめします。
Alkaline Phosphatase (BAP)
Q1 BAPはリン酸化したアミノ酸も脱リン酸化するのか?
A1 脱リン酸化します。BAPの活性は特異性が低く、ほとんど全てのリン酸モノエステル結合を同程度に加水分解します。ヒスチジノールホスフェイトに対する反応速度は、活性測定の基質である p -nitrophenyl phosphateを1として約0.9です。
Q2 制限酵素反応後に、直接BAPを添加して脱リン酸化することは可能か?
A2 制限酵素反応バッファー中でのBAP活性をBAP添付バッファーでの活性と比較した場合、HおよびKバッファーでは100%以上、L、M、T+BSAでも60%以上の相対活性を示しました。 したがって、5'突出末端の脱リン酸化反応など比較的簡単な脱リン酸化の場合には、制限酵素反応終了後に直接BAPを添加することでも脱リン酸化が可能と考えられます。
しかしながら、平滑末端や5'陥没末端の脱リン酸化、あるいは高い脱リン酸化効率が必要な系の場合には、制限酵素反応終了後にエタノール沈殿を行い、バッファー交換をしてから脱リン酸化を行うことをお勧めします。
しかしながら、平滑末端や5'陥没末端の脱リン酸化、あるいは高い脱リン酸化効率が必要な系の場合には、制限酵素反応終了後にエタノール沈殿を行い、バッファー交換をしてから脱リン酸化を行うことをお勧めします。
Alkaline Phosphatase (CIAP)
Q1 制限酵素処理後、バッファー置換せずにCIAPを使ってもよいか?
A1 最適条件に比べ数分の1から10分の1に効率が落ちます。しかし通常、大過剰量使用するので、DNAが5'突出末端なら問題ありません。5'陥没末端ならできるだけバッファー置換し、高めの温度(50℃くらい)で使用してください。
Q2 CIAPのイオン要求性は?
A2 Zn2+は不要(酵素タンパクと結合している)。Mg2+は0.5 mMで0 mMの場合の約3倍活性化します。
T4 DNA Polymerase
Q1 超音波処理して下図のようになったフラグメントはT4 Polで修復できるか?
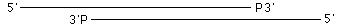
A1 できません。5'突出が脱リン酸化されたDNAなら基質となりますが、3'陥没末端にリン酸基を持つDNAには働きません。Mung Bean Nuclease(製品コード 2420)を作用させると平滑になります。
Q2 3'突出末端をT4 Polで削って平滑末端にしたいが、dNTPは必要か?
A2 必要です。ただし、必ずしも4種類必要でなく、平滑末端になったとき、3'側の一番端になる塩基を0.1 mM程度反応系に加えるだけでかまいません。T4 Pol 1 U、37℃、5 minの反応で充分でしょう。
Q3 Replacement synthesisの際、Tris-acetate系のバッファーで反応を行うと約40分でDNAがなくなってしまった。カタログに載っている活性測定用バッファー(Tris-HCl)で同じ実験をするとうまくいった。どういうことか?
A3 Tris-acetate系とTris-HCl系におけるT4 DNA Polymeraseのexonuclease活性を調べたところ、Tris-HCl系ではexonuclease活性がかなり抑えられることがわかりました。Tris-acetate系でreplacement synthesisを行うときには反応時間を短めにした方がよいでしょう。
Q4 KlenowとT4 DNA Polymeraseの差異は?
A4 T4 DNA Polymeraseの方が100~1000倍高い3'-exonuclease活性を持っています。またKlenowはT4 DNA PolymeraseほどDNAの二次構造の影響を受けません。したがってKlenowは、dideoxy法によるシークエンシング、DNAの3'陥没部分のfill-inや3'末端の標識に向いており、T4 DNA Polymeraseは3'末端の平滑化に向いています。
また、dNTP〔αS〕に対する活性も異なります。KlenowはdNTP〔αS〕を取り込みますが、exonuclease活性で削ることはできません。したがってDNAを一方向からのみ欠失させたい場合、KlenowとdNTP〔αS〕で一端を修復すれば、その末端はKlenowのexonuclease活性やBAL 31、Exo III等にも抵抗性になります。 しかし、T4 DNA PolymeraseはdNTP〔αS〕を取り込んでも削ってしまいます。
また、dNTP〔αS〕に対する活性も異なります。KlenowはdNTP〔αS〕を取り込みますが、exonuclease活性で削ることはできません。したがってDNAを一方向からのみ欠失させたい場合、KlenowとdNTP〔αS〕で一端を修復すれば、その末端はKlenowのexonuclease活性やBAL 31、Exo III等にも抵抗性になります。 しかし、T4 DNA PolymeraseはdNTP〔αS〕を取り込んでも削ってしまいます。
Klenow Fragment
Q1 KlenowとT4 DNA Polymeraseの差異は?
A1 T4 DNA Polymeraseの方が100~1000倍高い3'-exonuclease活性を持っています。またKlenowはT4 DNA PolymeraseほどDNAの二次構造の影響を受けません。したがってKlenowは、dideoxy法によるシーケンシング、DNAの3'陥没部分のfill-inや3'末端の標識に向いており、T4 DNA Polymeraseは3'末端の平滑化に向いています。
また、dNTP〔αS〕に対する活性も異なります。KlenowはdNTP〔αS〕を取り込みますが、exonuclease活性で削ることはできません。したがってDNAを一方向からのみ欠失させたい場合、KlenowとdNTP〔αS〕で一端を修復すれば、その末端はKlenowのexonuclease活性やBAL 31、Exo III等にも抵抗性になります。 しかし、T4 DNA PolymeraseはdNTP〔αS〕を取り込んでも削ってしまいます。
また、dNTP〔αS〕に対する活性も異なります。KlenowはdNTP〔αS〕を取り込みますが、exonuclease活性で削ることはできません。したがってDNAを一方向からのみ欠失させたい場合、KlenowとdNTP〔αS〕で一端を修復すれば、その末端はKlenowのexonuclease活性やBAL 31、Exo III等にも抵抗性になります。 しかし、T4 DNA PolymeraseはdNTP〔αS〕を取り込んでも削ってしまいます。
Q2 Klenowを16~22℃で使いたいが、このときの相対活性は?
A2 37℃のときに比べ、約1/2~1/3となります。fill-in反応等を低い温度で行う場合は長時間(10℃、1時間または0℃、3時間)反応させた方がよいでしょう(このときは1 μg DNAあたり3 Uくらいまで)。
Q3 DNAの修復に用いているが、DNAが少し分解されているようだ。
A3 Klenowは3'-exonuclease活性を持っている(一本鎖特異的とはいえ、相対的なものであり、二本鎖にも働く)ので1 μgに1 U以上用いると修復効果が低下します。1 μg DNAあたり0.1 U程度使うようにしてください。また、A-2のように低温で反応させる方がよいでしょう。
Q4 DNAの末端修復に用いているがライゲーション効率が悪い。
A4 Klenowは3'-末端をfill-inした後、1塩基余分に付加する傾向があります。突出した末端は3'-exonuclease活性で除去されますが、その効率が低いため完全には平滑化されません。平滑化のためにはT4 DNA Polymerase、またはDNA Blunting Kitをお勧めします。
Terminal Deoxynucleotidyl Transferase
Q1 カコジル酸入りのバッファーを用いて反応液を調製するとき、液が着色したり沈殿が生じたりする。
A1 H2O→カコジル酸→DTT→CoCl2(MgCl2、MnCl2)の順に加えていけば沈殿は生じません。
SP6 RNA Polymerase
Q1 RNAがうまく合成されないが?
A1 DNA、NTPの濃度、DTTをチェックしてください。DTTは活性の発現に重要です。
Q2 capped transcriptを効率よく作らせるにはどうすればよいか?
A2 capped analog/GTPの濃度比を10~20倍にしてください。たとえGTP濃度が50μMになっても完全鎖長のRNAの生成率は10分の1になることはありません。
Q3 capped analogのメチル化は反応効率に影響するか?
A3 ほとんどありません。
Q4 SP6 RNA Polymeraseを用いてRNAを合成する際、ビオチン化基質を取り込ませたいが可能か?
A4 RNA Polymeraseは基質特異性が高く、Biotin-UTPの取り込みはUTPの1/5以下です。
S1 Nuclease
Q1 末端平滑化の際のMung Bean Nuclease(MB)とのちがいは?
A1 MBは末端がGCだと削り残しやすく、S1は逆に削り込み(nibbling)を起こしやすいといわれています。DNAの末端によって使い分けるとよいでしょう(末端がGCrichならS1を使う。ただし削り込まないよう反応時間に注意。それ以外ならMBを使う)。
ただ、どちらの酵素も反応後T4 DNA Polymeraseで処理した方が、DNAの末端が平滑になる確率は高くなります。
ただ、どちらの酵素も反応後T4 DNA Polymeraseで処理した方が、DNAの末端が平滑になる確率は高くなります。
Mung Bean Nuclease
Q1 Mung Bean Nuclease(MB)を働かせたDNAがスメアしている。削り込んでいるのではないか?
A1 MBでは削り込みはほとんど起こりません。10~50 U/μg DNA、25℃、15 minで反応させてください。30 U、37℃、10 min以上では非特異的分解が起こる可能性があります。
また、DNAをMBで処理する前にExo IIIを使った場合、DNAにニックがあるとExo IIIがそこから働きます。ニックのないintactなDNAかどうか確認してください。
また、DNAをMBで処理する前にExo IIIを使った場合、DNAにニックがあるとExo IIIがそこから働きます。ニックのないintactなDNAかどうか確認してください。
Q2 MBとS1の使い分けは?
A2 MBはGCを残しやすく、S1は削り込み(nibbling)を起こしやすいといわれています。どちらもT4 DNA Polymeraseで末端を修復した方が平滑になる確率は高くなります。
Q3 MBをDeletion KitのMB Buffer中で使うとDNAがスメアした。
A3 キットのMB Bufferは、Exo III Buffer(pH8.0)と混ぜて使うよう設計してあるため、pHが5より低くなっています。そのため単独で使うと一本鎖DNAに対する特異性が低下し、dsDNAも分解されやすくなるのです。MBはpH5.0以上で使って下さい。
Q4 振とうおよび吸着により失活するか?
A4 しません。S1についても同じです。
BAL 31 Nuclease
Q1 BAL 31 Nucleaseの反応液組成には600 mMとかなり高濃度のNaClが入っているが、反応に影響はないのか?
A1 BAL 31は海洋細菌由来の酵素なので、高塩濃度でも充分活性を示します。60 mM、200 mM、600 mM NaClでexonuclease活性を比較したところ、あまり活性の差はなく、60 mMと600 mMでも2~3倍程度60 mMの方が高いだけです。60 mM NaClではニッキング活性によりendolyticな分解が起こっており、低塩濃度で使用するのは不適当です。600 mM NaClの方が分解速度が適当で、実用上は全く問題ないと思われます。
Q2 アデノウイルスDNAにBAL 31 Nucleaseを作用させたが分解されない。何故か?
A2 真核細胞由来のDNAにはBAL 31 Nuclease抵抗性のものがあります。塩濃度を下げた方がよいかも知れません。至適塩濃度はssDNAの場合600 mM~1 M NaClですが、dsDNAに対してはより低濃度の方が活性は高くなります。ただし200 mM以下の塩濃度ではdsDNAのアトランダムな分解が起こるので、200~600 mMの範囲で使ってください。なお、Sma IのようなGC richな制限酵素サイトは避けてください。
Q3 BAL 31 Nucleaseはニックからも働くか?
A3 程度は低いが働きます。
Q4 末端から数ヌクレオチドだけ除去したい。通常の条件では削りすぎて困るが?
A4 反応温度を下げる(20℃では30℃の約3分の1の活性)か、Exonuclease III とS1 Nucleaseの組合せをお使いください。
Exonuclease III
Q1 DNAにExo III を働かせた後、アガロース電気泳動するとスメアしている。非特異的なnucleaseがコンタミしているのでは?
A1 Exo III はニックにも作用します。DNAを制限酵素で切断するとき、過剰の酵素を用いたり、長時間反応させるとStar活性等によりDNAにニックが入ることがあります。
また、Exo III反応後、一本鎖部分を残したまま電気泳動しても正確なサイズは分かりません。MungBean Nuclease、またはS1 Nucleaseで一本鎖部分を削ってから電気泳動を行ってください。
また、Exo III反応後、一本鎖部分を残したまま電気泳動しても正確なサイズは分かりません。MungBean Nuclease、またはS1 Nucleaseで一本鎖部分を削ってから電気泳動を行ってください。
Ribonuclease Inhibitor
Q1 希釈した場合の安定性は?
A1 タンパク濃度の低下により失活しやすくなります。また、撹拌による失活も大きくなります。できるだけ原液のまま反応系に添加することをお勧めします。
Q2 熱安定性は?
A2
| 55℃ | 問題ありません。 |
| 65℃ | RNaseAの力価減少のため、inhibitorの正確なassayができません。 ただし、活性は維持されているようなので使用する意義はあります。 |
| 75℃ | RNaseAの活性がなくなるため、assay不能となります。 |
Q3 RNaseHに作用するか?
A3 影響しません。
Q4 DTTの代わりに2-メルカプトエタノール(2-ME)ではどうか?
A4 酸化還元電位はDTT>2-ME(10倍くらい)です。このため40~50 mMの2-MEが必要です。この場合、inhibitorの活性は変化ありませんが、他の反応への影響が心配されます。