
様々な生物種のRNAから方向性の情報をもつNGS用ライブラリー調製が可能
- 99%以上の精度でトランスクリプトをカバーするライブラリー調製が可能
- わずか100 pgのrRNA除去済みRNAから質の高いRNA-Seq解析データを取得可能
- PCR増幅ステップでインデックスとアダプターを組み込むシームレスなワークフロー
- 4時間以内に方向性情報を保持したRNA-Seqライブラリーを調製
- Unique Dual Index Kit(別売り)と組み合わせることにより、最大384サンプルのマルチプレックス解析に対応可能
注: SMART-Seq Total RNA Mid Inputは、SMARTer Stranded RNA-Seq Kit(製品コード 634838)、SMARTer Stranded RNA-Seq Kit HT(製品コード 634862:終売)、およびSMARTer Stranded Total RNA Sample Prep Kit - Low Input Mammalian(製品コード 634861:終売)のリニューアル製品です。基本性能、プロトコールに変更はありませんが、インデックスが別売りとなっていますので、必ず別途ご購入ください。(Unique Dual Index Kit参照)
新製品と既存製品の対応表はこちら(TB USA社のサイト)
新製品と既存製品の対応表はこちら(TB USA社のサイト)
※本キットにはIndexが含まれていません。Unique Dual Index Kit(製品コード 634752/634753/634754/634755/634756)を別途ご購入ください。
※シーケンスを行う際には、サンプルにControl DNA(PhiX)を加えて解析することをおすすめします。
※シーケンスを行う際には、サンプルにControl DNA(PhiX)を加えて解析することをおすすめします。
カートにいれる
WEB会員ログインが必要です
製品説明
SMART-Seq Total RNA Mid Inputは、rRNAを除去した100 pg~100 ngのRNAからインデックスを付加したイルミナ社次世代シーケンサー対応のライブラリーを調製するキットです。本キットはランダムプライマーによるcDNA合成を行うため、真核生物および原核生物のRNAから、コーディングRNAとノンコーディングRNAの両方のトランスクリプトを99%以上の精度でカバーしたライブラリーの調製が可能です。イルミナ社次世代シーケンサー対応のインデックスとアダプターを組み込むステップがワークフロー中に含まれているため、別途、ライブラリー調製キットを用意する必要はなく、別売りのUnique Dual Index Kit(製品コード 634752~634756)と組み合わせることにより、最大384サンプルのマルチプレックス解析に対応可能です。
なお、ヒト、マウス、ラットtotal RNA(10~100 ng)サンプルからのrRNAの除去には、RiboGone-Mammalian(製品コード 634847)の使用をおすすめします。
なお、ヒト、マウス、ラットtotal RNA(10~100 ng)サンプルからのrRNAの除去には、RiboGone-Mammalian(製品コード 634847)の使用をおすすめします。
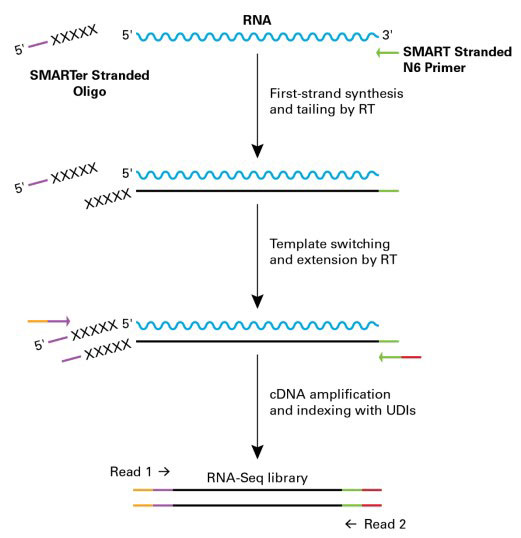
図1. SMART-Seq Total RNA Mid Inputを用いたライブラリー調製の概要
※ 以下のデータは、SMART-Seq Total RNA Mid Inputを同じ原理・性能のSMARTer Stranded RNA-Seq Kit(製品コード 634838)で得たものを示しています。
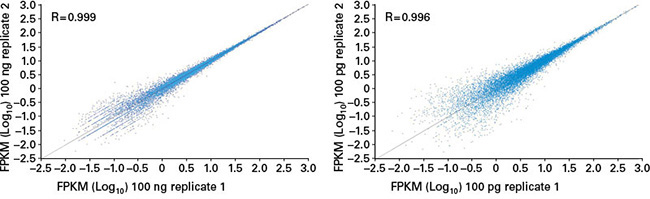
図2. テクニカルレプリケート間での遺伝子発現量の比較
RNA 100 ngまたは100 pgを用いた繰返し実験において、再現性の高いデータが得られた。
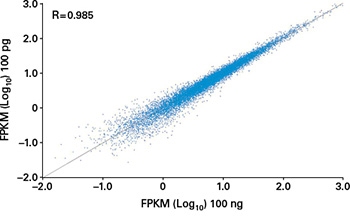
図3. インプット量を変えたライブラリー間での遺伝子発現量の比較
ヒト脳poly(A)+ RNA 100 ngおよび100 pgから調製したcDNAライブラリーの発現解析結果(FPKM:fragments per kilobase of exon per million reads mapped)を示す。
RNA量に1,000倍差のあるサンプル間の比較においても、高い再現性と同様の感度が示された。
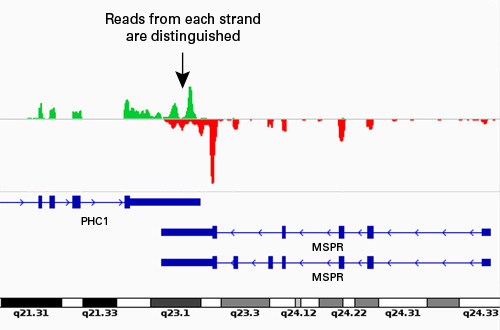
図4. 12種類のインデックスを用いたcDNAライブラリーのイルミナ社HiSeqシーケンサーによる解析
ゲノムDNAの各strandから転写されたリードは正確に区別されるため、転写産物の発現定量や正確なアノテーションが可能である。RNA-Seqリードの99%は正しいDNA鎖へ正確にマッピングされた。
表1. RNA量100 pgを用いたシーケンス解析
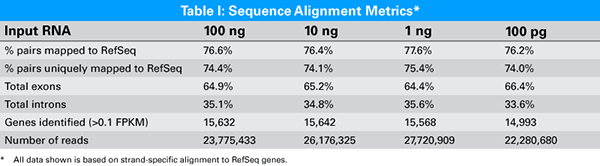
わずか100 pgのインプットからでも転写されたDNA鎖を正確に特定することができ、また約15,000種類の遺伝子を検出することが可能であった。
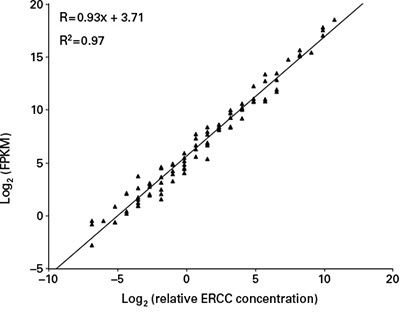
図5. ERCC解析での高い再現性
事前に取得した複数のERCC(External RNA Control Consortium)データセットの相対的転写産物量に、SMARTer Stranded RNA-Seq Kit(製品コード 634836)で得られたFPKMをマップした。92個のERCCセットがスロープ=0.934、R2=0.9725でマップされた。

図6. cDNAライブラリーの収量および純度
ヒトpoly(A)+ RNA 100 ng~100 pgから調製されたcDNAライブラリーの収量、純度をAgilent 2100 Bioanalyzerを用いて分析した。cDNAの増幅に使用したサイクル数も示す。cDNAライブラリーはSeqAmp DNA Polymerase(本キットに含まれる)により増幅した。
RNAのインプット量にかかわらず、一貫した収量と純度でライブラリー調製が可能であることが示された。
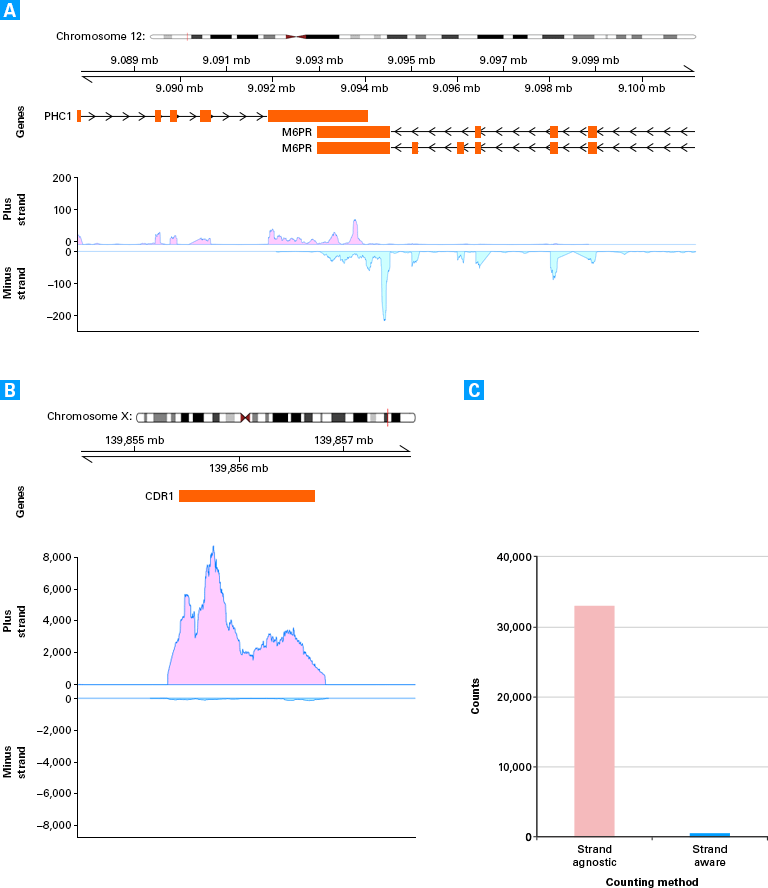
パネルA:ヒト脳poly(A)+ RNA cDNA libraryのリードをヒトゲノムDNAに対してマッピングした。
SMARTer Strandedメソッドにより、PHC1とM6PRのリードが重複する場合でも、シーケンスリードを正しくマッピングすることができた。
パネル B:CDR1 locusのStrand特異的カバレージ。ほぼすべてのリードは、アノテーションされたトランスクリプトに対してアンチセンス側の配列を示した。
Hansen, T. B. et al. (2011) EMBO J. 30(21):4414–4422.
パネル C:strand-agnostic法またはstrand-aware法によって得られたCDR1遺伝子カウントの比較
内容
SMART-Seq Total RNA Mid Input(製品コード 635048/635049/635050)
- Box 1
-
- Control Mouse Liver Total RNA (1 μg/μl)
- SMARTer Stranded Oligonucleotide (12 μM)
- Box2
-
- SMART Stranded N6 Primer (12 μM)
- First-Strand Buffer (RNase-Free) (5X)
- dNTP Mix (dATP, dCTP, dGTP, and dTTP, each at 10 mM)
- Dithiothreitol (DTT; 100 mM)
- SMARTScribe Reverse Transcriptase (100 U/μl)
- Nuclease-Free Water
- RNase Inhibitor (40 U/μl)
- Stranded Elution Buffer
- SeqAmp DNA Polymerase
- SeqAmp PCR Buffer (2X)
保存
Box1:-70℃
Box2:-20℃
(Stranded Elution Buffer:融解後は室温保存)
Box2:-20℃
(Stranded Elution Buffer:融解後は室温保存)
本製品以外に必要な試薬、機器(主なもの)
下記の製品は本キット中に含まれません。下記の製品はユーザーマニュアルに記載のProtocolで使用できることが確認されています。
Index Kit
PCR増幅およびバリデーション
Beads精製
その他
Index Kit
- Unique Dual Index Kit (1-24)(製品コード 634756)
UDI No. U001-U024 - Unique Dual Index Kit (1-96)(製品コード 634752)
UDI No, U001-U096 - Unique Dual Index Kit (97-192)(製品コード 634753)
UDI No, U097-U192 - Unique Dual Index Kit (193-288)(製品コード 634754)
UDI No, U193-U288 - Unique Dual Index Kit (289-384)(製品コード 634755)
UDI No, U289-U384
PCR増幅およびバリデーション
- First-strand cDNA合成専用のPCRサーマルサイクラー
- Agilent High Sensitivity DNA Kit (Agilent Technologies社, Cat. No. 5067-4626)
- Nuclease-free, PCR-grade, thin-wall PCR strips (0.2 ml PCR 8-tube strip; Thermo Fisher Scientific社, Cat. No. AB0264) or similar nuclease-free, PCR-grade, thin-wall PCR tubes, strips, or 96-well plates
- Nuclease-free low-adhesion 1.5 ml tubes(USA Scientific社 Cat No. 1415-2600)または、DNA LoBind tubes(Eppendorf社 Cat. No. 022431021)
Beads精製
- NucleoMag NGS Clean-up and Size Select(5 ml size:製品コード 744970.5;50 ml size:製品コード 744970.50;500 ml size:製品コード 744970.500)
※ NucleoMagの代わりにAMPure XP PCR purification kit(Beckman Coulter社 5 ml size:Cat. No. A63880;60 ml size:Cat. No. A63881)も使用可能 - 80%エタノール(用事調製)
- Magnetic Separation Device
0.2 mlチューブ用マグネットスタンド
96-well plate:Magnetic Stand 96(Thermo Fisher Scientific社 Cat. No. AM10027) - 96-well plate用低速遠心機
その他
- シングルチャンネルピペット:10 μl、20 μlおよび200 μl
- 8連、あるいは12連チャンネルピペット:10 μl
- フィルターピペットチップ:10 μl、20 μl、および200 μl
- 1.5 mlチューブ用微量遠心機
- 0.2 mlチューブまたはstrip用微量遠心機
この製品を見た人は、
こんな製品も見ています
-

SMART-Seq® mRNA / SMART-Seq® mRNA LP
イルミナ社NGS装置用の超微量mRNA-Seq用ライブラリー調製キット
-

SMART-Seq® Total RNA High Input (RiboGone™ Mammalian)
total RNA 100 ng~1 μgからイルミナ社NGS用ライブラリーを迅速に調製
-

SMART-Seq® Total RNA Single Cell Library Prep with ZapR® Depletion
シングルセルまたはtotal RNA 10 pgからの方向性情報を持ったtotal RNA-Seq解析
-

SMART-Seq® Stranded Kit
シングルセルまたはtotal RNA 10 pgからの方向性情報を持ったtotal RNA-Seq解析
-
次世代シーケンス(NGS)関連試薬選択ガイド RNAシーケンス
-

SMART-Seq® Total RNA Library Prep with ZapR® Depletion
微量total RNA(250 pg~1 μg)から方向性情報を持ったNGS用ライブラリー調製が可能
- 注意事項
- 本ページの製品はすべて研究用として販売しております。ヒト、動物への医療、臨床診断用には使用しないようご注意ください。また、食品、化粧品、家庭用品等として使用しないでください。
- タカラバイオの承認を得ずに製品の再販・譲渡、再販・譲渡のための改変、商用製品の製造に使用することは禁止されています。
- タカラバイオ製品に関連するライセンス・パテントについては、ライセンスマークをクリックして内容をご確認ください。
また、他メーカーの製品に関するライセンス・パテントについては、各メーカーのウェブサイトまたはメーカー発行のカタログ等でご確認ください。 - ウェブサイトに掲載している会社名および商品名などは、各社の商号、または登録済みもしくは未登録の商標であり、これらは各所有者に帰属します。