
プロトコール C. 高力価組換えアデノウイルスの作製および確認、D. 力価測定、E. 目的細胞へのアデノウイルスの感染
C. 高力価組換えアデノウイルス液の作製
- コラーゲンコート25 cm2フラスコに70~100%コンフルエントまで培養した293細胞を用意する。
- B-2.の解析で選択した目的のウイルス株の2次ウイルス液を感染させる。ウイルス液15 μlと5%FCS-DMEM培地0.5 mlを、培地を除いたフラスコに静かに加える。
- フラスコをシーソーのように数回、ゆっくりと振とうさせ、ウイルス液をすべての細胞にいきわたらせ感染を行う。この操作を15~20分ごとに3~4回行う。この間、細胞はCO2インキュベーター(37℃、5%CO2)においておく。
- 1時間の感染後、5%FCS-DMEM培地 4.5 mlを加える。
- 3~4日後、すべての細胞が変性したら、培地ごと細胞を無菌的に滅菌チューブに回収し、凍結融解または密閉型ソニケーターで破砕してウイルスを遊離させる。
* 容量が大きい場合密閉型ソニケーターを使用されることをお勧めします。なお、開放型のソニケーターはエアロゾルが発生するので使用しないで下さい。 - 3,000 rpm、10分、4℃で遠心し、上清を回収する。1 mlずつ5本のシーリングキャップ付マイクロチューブに分注し、ドライアイスで急凍して‐80℃で保存する(3次ウイルス液)。
- 3次ウイルス液50 μlと5%FCS-DMEM 2 mlを、コラーゲンコート75 cm2フラスコで70~100%コンフルエントまで培養した293細胞に、C-3.と同様に感染させる。
- 1時間の感染後、5%FCS-DMEM 13 mlを加える。
- 3~4日後、すべての細胞が変性したら、3次ウイルス液と同様にウイルス液を調製する。
- シーリングキャップ付マイクロチューブに1 mlずつ15本に分注し、ドライアイスで急凍して‐80℃で保存する(4次ウイルス液)。4次ウイルス液は、実際に実験に用いるもので109 PFU/ml程度の高力価となる。
- 4次ウイルス液は最初の使用時に1本を0.1 mlずつ分注して、ドライアイスで急凍後 ‐80℃に保存する(working stock)。凍結融解はなるべく避ける。
- 4次ウイルス液5 μlを、24ウェルプレート1ウェルの293細胞に感染させ、増殖したウイルスDNAの制限酵素パターンをB-2.の方法で確認する。
* もし欠失ウイルスあるいは親ウイルスとの混合物であることが疑われたら、2次ウイルスの段階で既にわずかに混在していたウイルスがその増殖が速いために見えてきた可能性があります。全ての3次、4次ウイルスを破棄し、別の2次ウイルスから改めてウイルス液の調製をやりなおすか、その1次ウイルス液から限界希釈法により目的ウイルスを純化して下さい。
* 実際に感染実験に使用するウイルス液は、継代ごとに欠失ウイルスや親ウイルスが生じていないことを確認してから用いることをお勧めします。
D. 力価測定[50% Tissue Culture Infectious Dose (TCID50) 法]
力価の測定には、寒天培地上でのプラークの形成を観察する方法(プラーク形成法)を用いるのが一般的であるが、この方法と以下に示したTCID50法との結果はよく一致します。
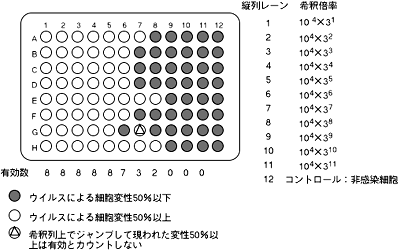 図4 50% Tissue Culture Infectious Dose(TCID50)測定法の実際
図4 50% Tissue Culture Infectious Dose(TCID50)測定法の実際
Karberの式より
TCID50=(1列目の希釈倍率)×(希釈倍率)Σ-0.5
ただし、Σ=各希釈段階における(変性50%以上のウェル数)/(検体数)の総和
上記の例では
Σ=8/8+8/8+8/8+8/8+8/8+7/8+3/8+2/8=6.5
したがって、TCID50=3×104×36.5-0.5=2.2×107
使用したウイルス液は50 μlなので、TCID50=PFUとすると、ウイルス原液の力価は
2.2×107×1 ml÷0.05 ml=4.4×108 (PFU/ml)
- 10 cm2細胞培養用シャーレ1枚に70~100%コンフルエントまで培養した293細胞を用意する。
- ウイルス液を5%FCS-DMEMで10倍ずつ段階希釈し、104倍希釈ウイルス液を用意する。例えば、ウイルス液0.1 mlに5%FCS-DMEM 0.9 mlを加えて希釈する。
- コラーゲンコート96ウエルプレート1枚の全てのウエルに50 μlずつ5% FCS添加DMEMを入れる。
- 第1列目に104倍希釈した組換えウイルスを25 μlずつ加える。
- 8チャンネルマルチピペッターを用いて25 μlを2列目のウエルに移す。
以下同じ操作を11列目まで繰り返し最後の25 μlを捨てる。結果として3nの段階希釈液を104×311の希釈段階まで作製することができる。12列目は非感染細胞のコントロールとする。チップは1列ごとに替える。 - 培養しておいた293細胞(D-1.)を6 mlの5% FCS添加DMEMに懸濁する。
- D-6.の細胞懸濁液を50 μlずつ各ウエルに加える。
- 4~5日後と7~8日後に各ウェルに50 μlずつ10% FCS添加DMEMを穏やかに加える。
- 11-13日後に細胞変性の終末点を顕微鏡で判定する。
* ウイルスが存在しているウェルの細胞は変性を起こして剥がれます(付録1-6.)。14日まで細胞を維持できれば判定は容易ですが、細胞が傷むと難しくなります。 - Karberの式を用いて統計学的に50%細胞変性終末点(TCID50)を計算する。293細胞を用いて本法で算出したTCID50の値と、プラーク形成法で求めたPFUの値とはよく一致する。実際の例を図4に示す。
TCID50=(1列目の希釈率)×(希釈率)Σ-0.5
ただし Σ = 各希釈段階における(変性ウエル数)/(検体数)の総和
図4 50% Tissue Culture Infectious Dose(TCID50)測定法の実際Karberの式より
TCID50=(1列目の希釈倍率)×(希釈倍率)Σ-0.5
ただし、Σ=各希釈段階における(変性50%以上のウェル数)/(検体数)の総和
上記の例では
Σ=8/8+8/8+8/8+8/8+8/8+7/8+3/8+2/8=6.5
したがって、TCID50=3×104×36.5-0.5=2.2×107
使用したウイルス液は50 μlなので、TCID50=PFUとすると、ウイルス原液の力価は
2.2×107×1 ml÷0.05 ml=4.4×108 (PFU/ml)
E. 目的細胞へのウイルスの感染
接着細胞に感染させる場合の一般的な方法を示します。浮遊細胞の場合は細胞を遠心して集めウイルス液に懸濁し、同様の操作を行います。
- 70~100%コンフルエントまで培養した目的細胞を1種類のウイルスあたり5ウェル以上(以下に示すウイルスの希釈の数だけ)用意する。
- 培地を除く。培地の血清がFCSでない場合(例えばCS)は、無血清の培地で2度細胞を洗い、培地を除く。
- 4次ウイルスの原液と、これを無血清またはFCS添加の培地で3倍、10倍、20倍および40倍に希釈したウイルス希釈液を、目安として96ウェルプレートで30~40 μl/ウェル、24ウェルプレートで50~70 μl/ウェル、10 cmシャーレで100~200 μl程度加える。
* 目的の遺伝子を効率よく発現させる条件は、目的細胞の性質によって異なります。まずは、数段階のウイルス希釈液を用いて至適条件を検討する必要があります。 - プレートをシーソーのように数回、ゆっくり振とうし、ウイルス液を全ての細胞にいきわたらせ、ウイルスを細胞に感染させる。この操作を15~20分ごとに3~4回行う。この間、細胞はCO2インキュベーター(37℃、5% CO2)においておく。
- 1時間の感染後、培地を適当量加えて培養する。
* 感染時間は通常1時間、長くても2時間程度で充分である。 - 0、1、2、3日後に蛍光抗体法やウェスタン法など、適当な方法で目的とするタンパク質の発現を検出する。